Allelic-specific methylation profiles in a genetic isolate
School of Medical Sciences, Institute of Health and Biomedical Innovation, Queensland University of Technology, Brisbane, Australia
AB3ACBS 2016 - QUT, Brisbane, Australia
1st-2nd November 2016

My presentation available online
![]()

http://sirselim.github.io/presentations.html
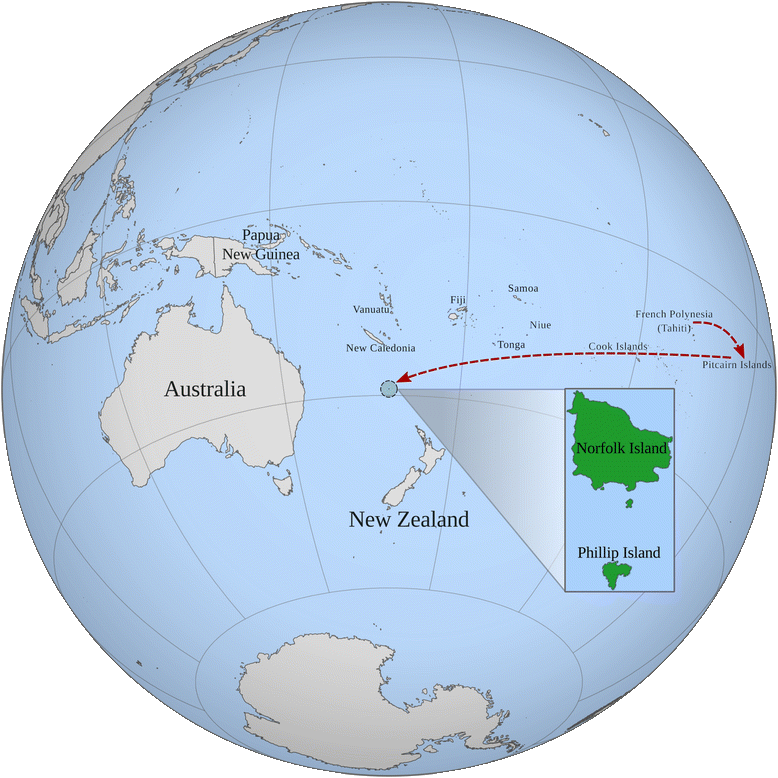
Norfolk Island
...and the Norfolk Island Health Study...
https://github.com/gringer/bioinfscripts/blob/master/perlshaper.pl

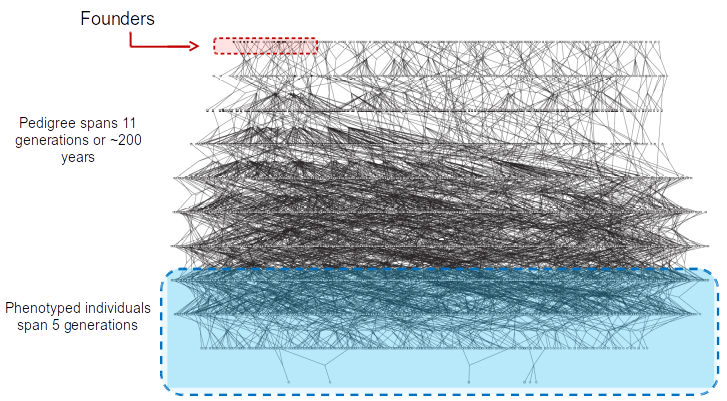
~6000 member pedigree
Macgregor S et al.,: Legacy of mutiny on the Bounty: founder effect and admixture on Norfolk Island. Eur J Hum Genet. 2010; 18: 67–72.
Mitochondrial Ancestry

40% of current population haplogroup B4a1a[...]
Benton MC et al.,: “Mutiny on the Bounty”: the genetic history of Norfolk Island reveals extreme gender-biased admixture. Investigative Genetics 2015, 6:11.
Allele-specific methylation
(very recent data)Other contributors: Rod Lea, Donia Macartney-Coxson, Nicole White, Daniel Kennedy, Heidi Sutherland, Larisa Haupt, Kerrie Mengersen, Lyn Griffiths



Allele-specific methylation
Allele-specific methylation (ASM): same cytosine is differentially methylated on the two alleles of a diploid organism
ASM is a major mechanism of genomic imprinting (aberrations can lead to disease)
Identification of allele-specific methylation profiles across generations
Proof of principle pilot study
- measuring genome-wide allele-specific methylation (ASM)
- NGS bisulphite sequencing
- SeqCap Epi CpGiant (Illumina HiSeq)
- collected data for 24 NI individuals
- comprising a close 3 generation pedigree
- currently generating data for another ~90 samples
- Fully customised QC and analysis pipeline:*
- fastqc, trimgalore
- bismark, sambamba, picard tools
- PileOMeth, R and methylkit
- parallel processing enabled for local and remote machines
- filtered at minimum 10 counts
*once wrangled into shape scripts will be accessible via GitHub
Initial metrics
- coverage ~40x ave
- lowest number of CpGs for a given sample is 2.67M
- highest number of CpGs for a given sample is 7.52M
- the average across all samples is 3.48M
- on-target mapping rate across the 24 samples >95% average
~1.12M CpG sites in common across the 24 samples with at least 10 times coverage and on-target
QC

QC
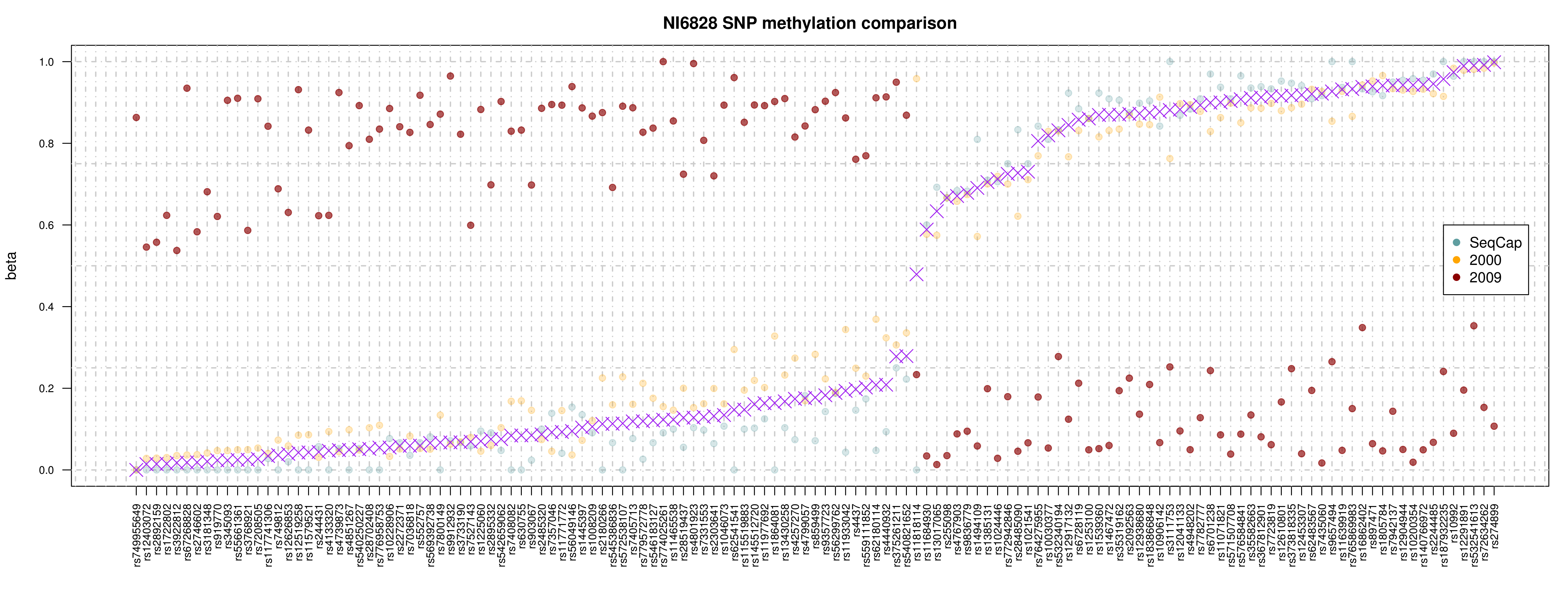
Allelic-specific methylation
- for an initial look-see used methpipe suite [1]
- estimates ASM at each CpG site based on ratios of methylation
- uses CpG pairs (assumes 'linkage')
- moving forward we want to use read data:
- assign parent of origin and then infer ASM
- gives the actual info rather than just knowing where an ASM event is
- promising method recently published for mice ASM [2]
[1]Fang F, et al.,: Genomic landscape of human allele-specific DNA methylation. PNAS, 2012; 109: 7332–7.
[2]Krueger F, et al.,: SNPsplit: Allele-specific splitting of alignments between genomes with known SNP genotypes. F1000Research, 2016; 5: 1479.
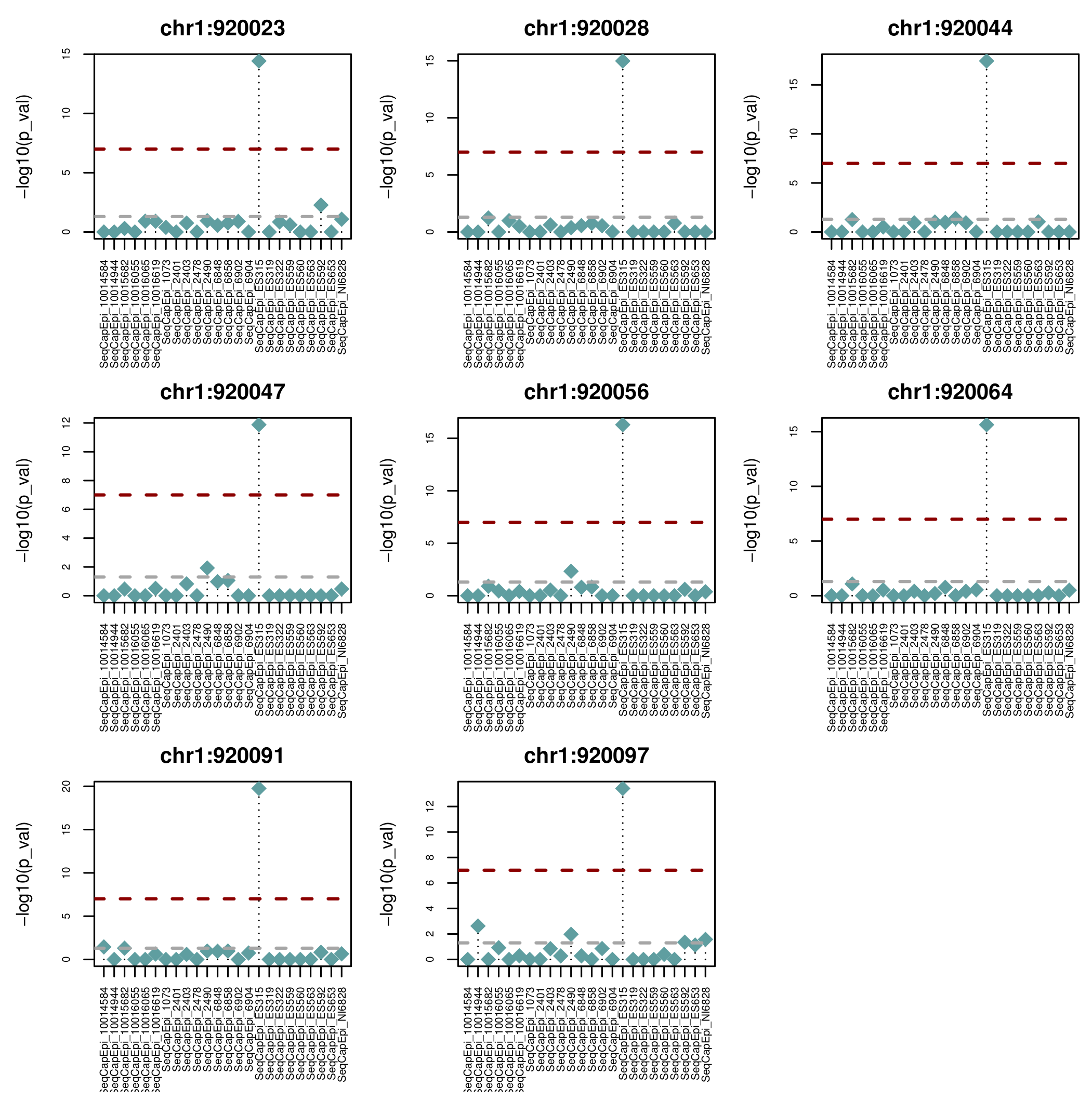
ASM regions
- Using a custom clustering method we identified ~1800 ASM regions (AMRs) conserved within the pedigree
- Many of these AMRs map to known and predicted imprinted genes
Visualising ASM
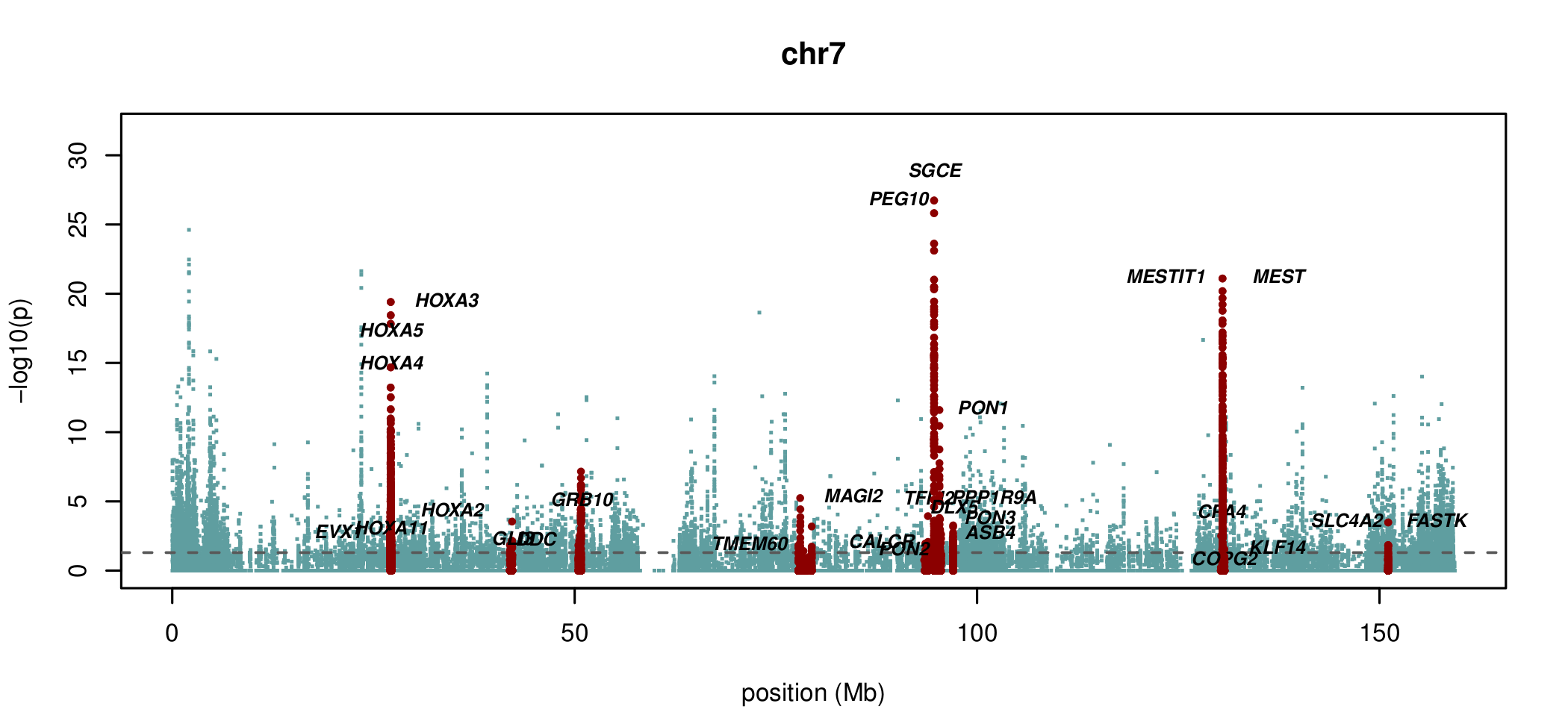
Documented and predicted human imprinted loci are displayed in darkred.
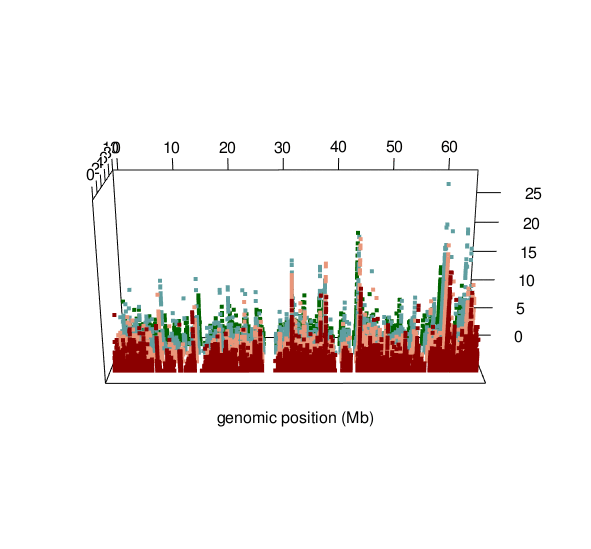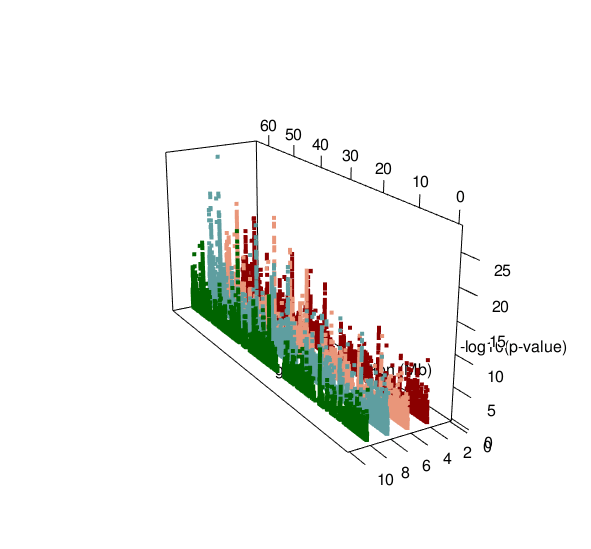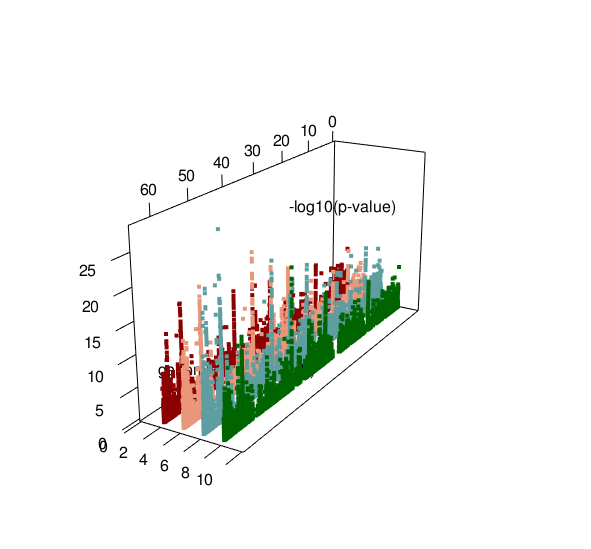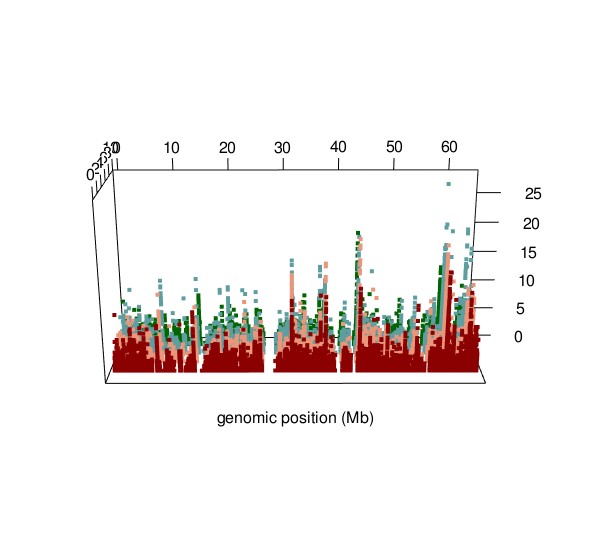
ASM plot of chromosome 20 for a nuclear family (father, mother, son, daughter)
Presence of SNPs
- 1,127,867 total methylation sites
- 444,330 have a SNP present on C/G (39.4%)
...however...
Considering MAF:
- 231,452 SNPs have recorded MAF info
- 12,670 SNPs >= 0.05 MAF
- 26,935 SNPs >= 0.01 MAF
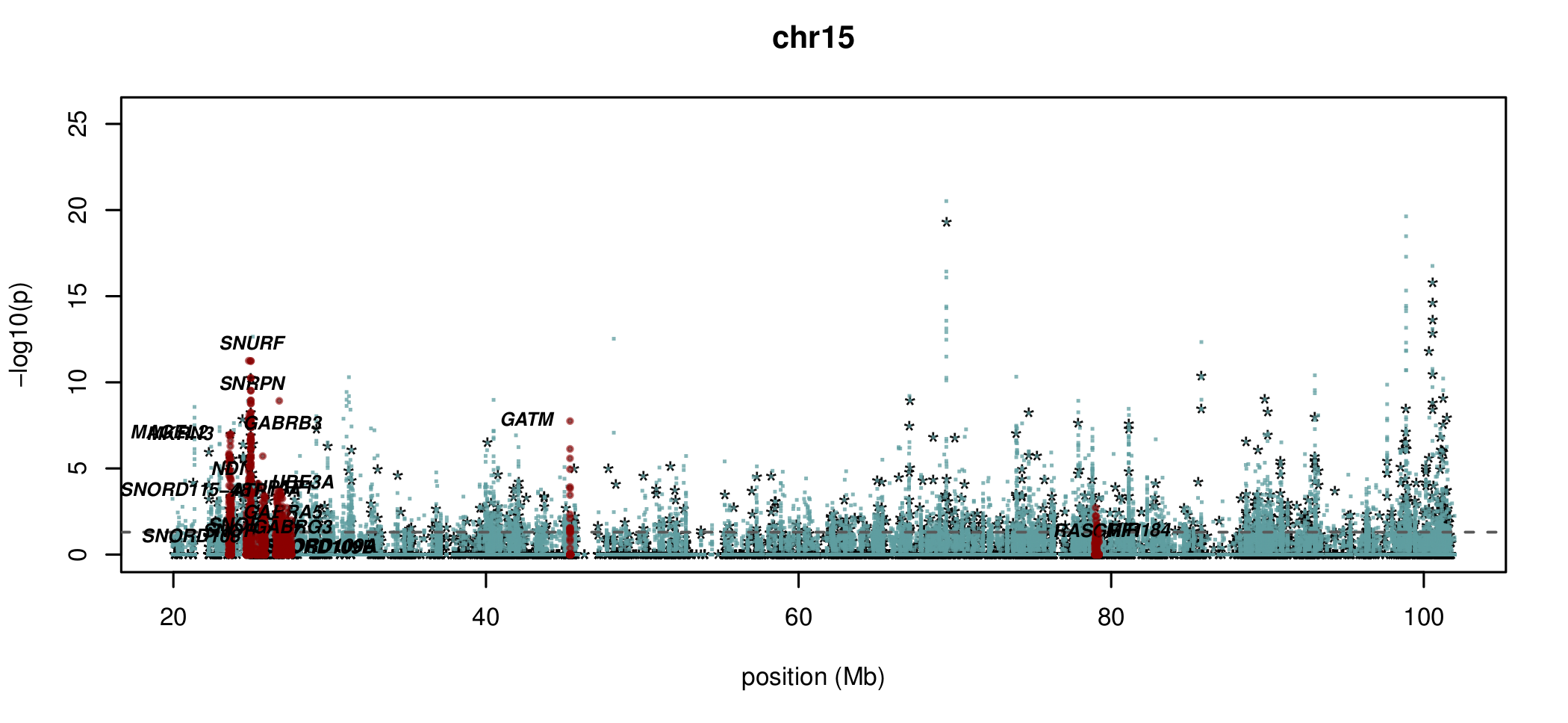
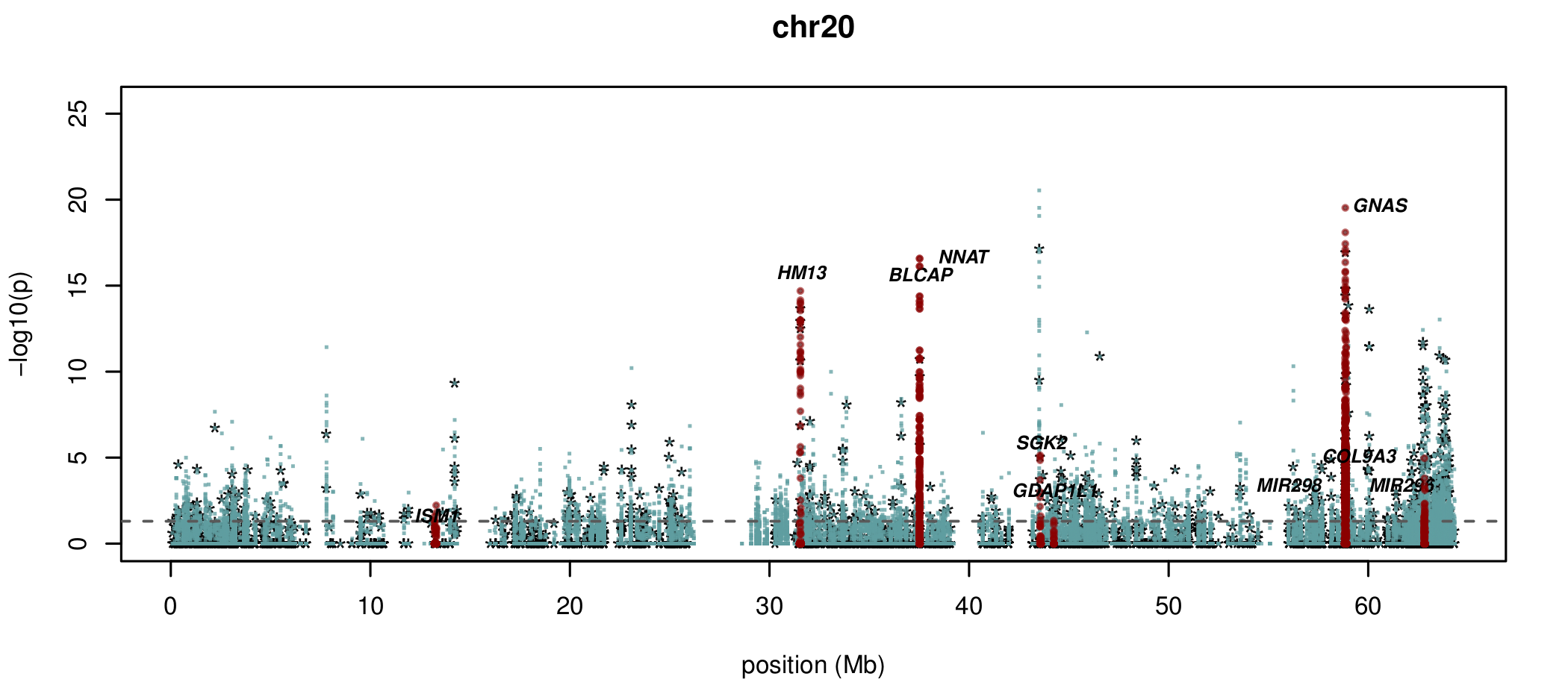
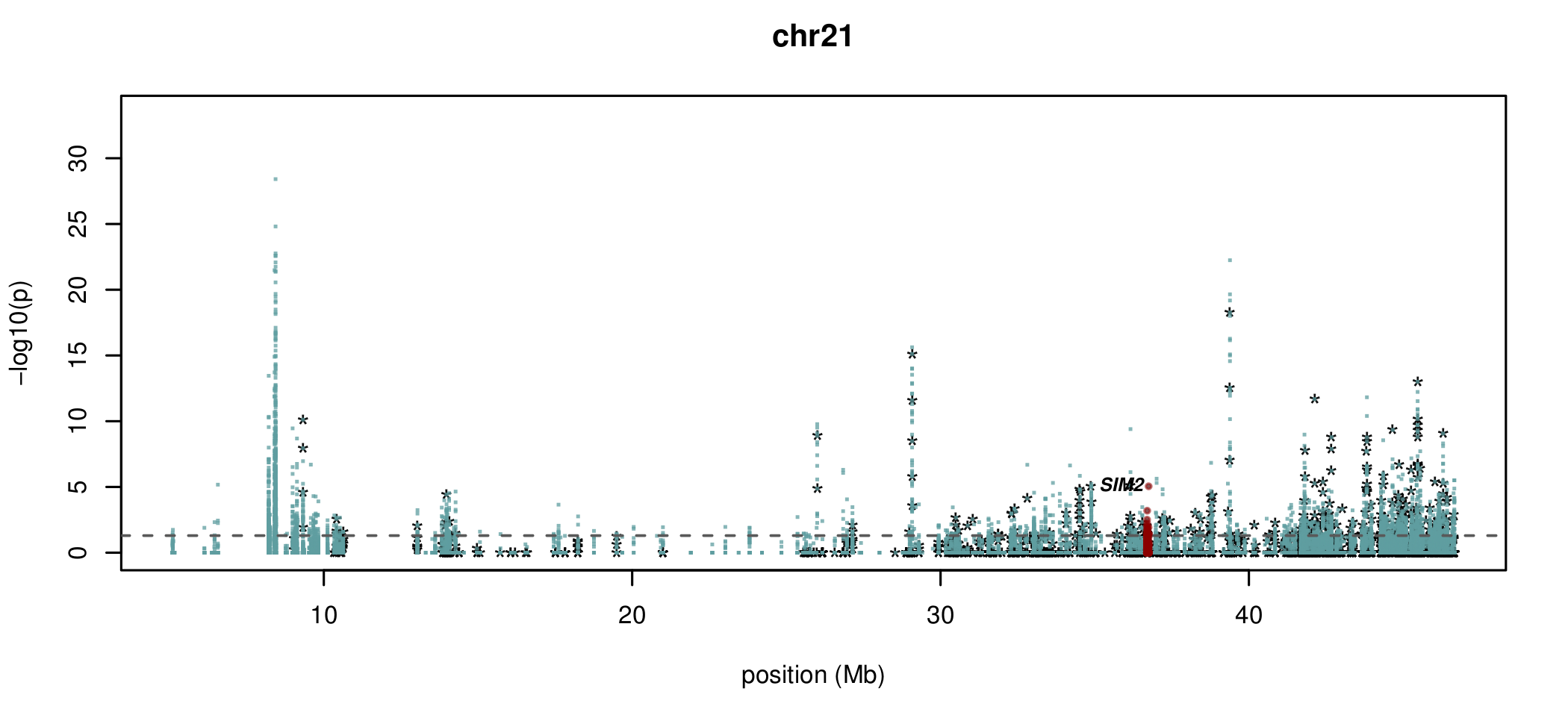

What about sites which aren't conserved?
Nice sanity check of method (X chromosome)
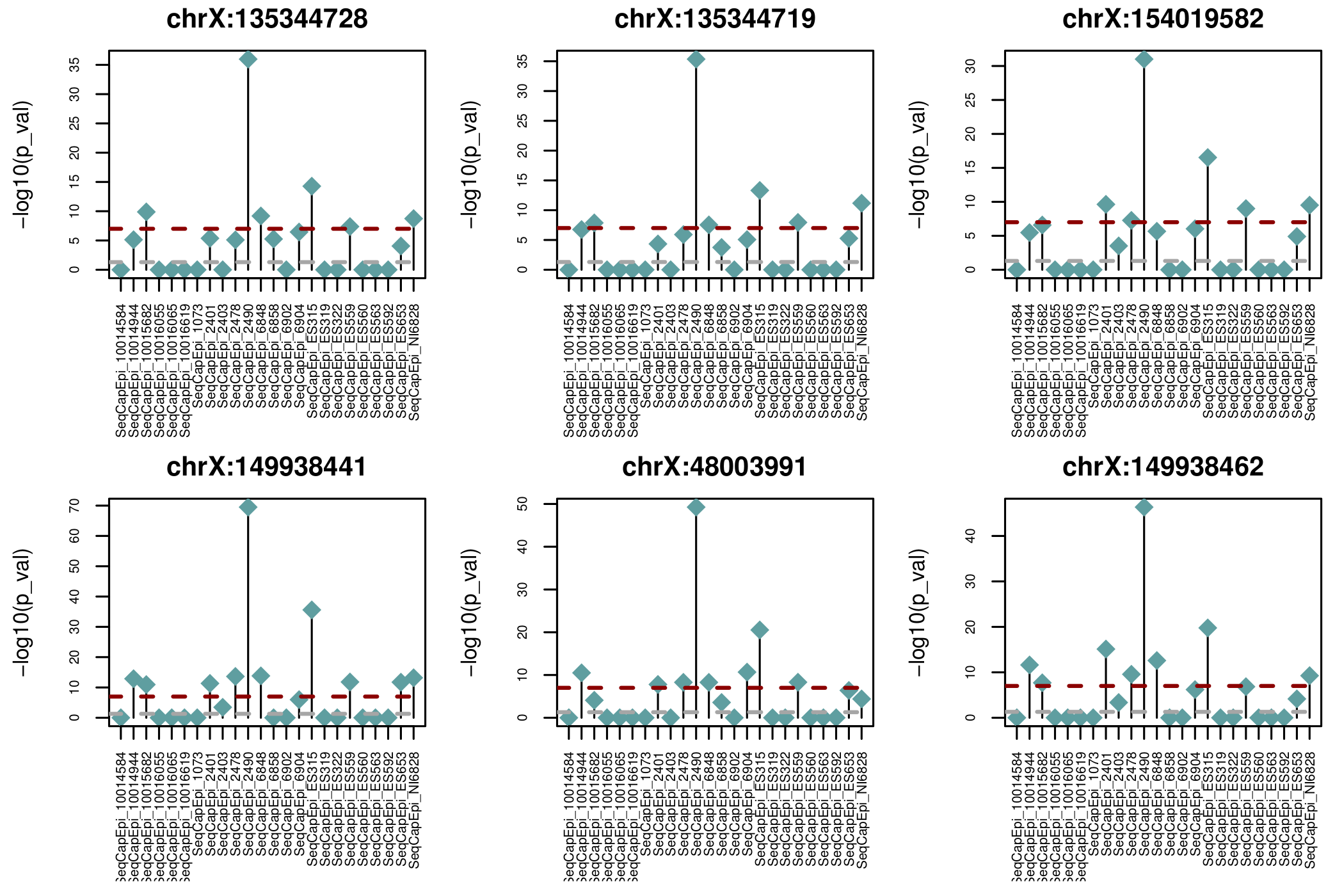
Epigenetic inheritance
Next thing is to move onto modeling the inheritance......with the help of our brilliant Bayesian statisticians!
Summary
- expanding upon our multi-layered NI data
- custom pipeline developed to interrogate bisulfite sequence data
- initial ASM region and imprinting loci identification
- potential to identify previously unreported imprinted loci
- in a position to start inheritance modeling
?
acknowledgments
| QUT | STDOI @ UTRGV |
NZ collaborators |
|---|---|---|
| Lyn Griffiths |
John Blangero |
Donia Macartney-Coxson (ESR) |
| Rod Lea |
Joanne Curran |
David Eccles (Gringene Bioinformatics) |
| Larisa Haupt |
Harald Goring |
Geoff Chambers (VUW) |
| Heidi Sutherland |
||
| Michelle Hanna |
||
| ...the rest of the GRC IHBI lab group | ||
| Nicole White | ||
| Daniel Kennedy | ||
| Kerrie Mengersen |
| Garvan Institute of Medical Research / Kinghorn Centre for Clinical Genomics | Melanie Carless (Texas Biomedical Research Institute) | NHMRC |
Claire Bellis (Genome Institute of Singapore) |
||
Greg Gibson (Georgia Institute of Technology, USA) |
The people of Norfolk Island who who volunteered for this study.
